固态电解质
固态电解质是通过一种原位熔化反应,在电解质颗粒表面生成共价键配位,来解决固态电池的氧化稳定性差和枝晶的问题。这种配位通过共价键合阴离子上的局部高浓度电子,从热力学角度关闭了阴离子氧化分解过程中的电子交换,并从动力学角度阻止了电解质颗粒表面的电子渗流;这种现象导致了一个前所未有的电压窗口(0 ~ 10 V),其峰值氧化电流比 25 ℃ 时的同类产物低 370 倍,电子电导率低 3 个数量级。该配位体可作为粘结剂粘结电解质颗粒,其杨氏模量高达 208.45 GPa;该模量是同类电解质的两倍,可适应锂沉积和剥离过程中的持续应力应变释放。凭借这些优点,该电解质在 25 ℃ 时的临界电流密度达到了破纪录的 21.65 mA cm^-2^(是锂离子固态电解质最佳报告数据的两倍),在10.83 mA cm^-2^下 稳定循环6000 小时,在10V下稳定1000小时。工作温度窗口宽达 -30 ℃ 至 150 ℃。* 开发的钴酸锂电池在高压下表现出卓越的可逆性。该研究结果为固态电解质的氧化稳定性和枝晶抑制指明了方向,为高压锂电池的发展带来了巨大**的进步。
 微软雅黑; font-size: 16px; letter-spacing: 0.5px;" />
微软雅黑; font-size: 16px; letter-spacing: 0.5px;" />基本介绍 编辑本段
在全固态电池(ASSBs)中使用固态电解质(SEs)是一项非常有前途的技术,由于其固有的安全性和稳定性,可以克服液态电解质的缺点,为发现具有超高能量密度的新型电池化学物质提供了机会,如金属锂电池(例如,金属锂阳极与高电压阴极配对)。然而,大多数无机 SE 都面临着氧化稳定性差(2 ~ 5 V 对 Li^+^/Li)和枝晶晶形成的问题,室温(RT)下的临界电流密度(CCD)小于 11 mA cm^-2^,这极大地阻碍了它们的应用。
因此,阴离子工程,如在 SE 中形成电负性更高的阴离子,可显著提高 SE 的氧化稳定性,从而在报道的最佳研究中实现 0 至 5 V 的大电压窗口。然而,迄今为止还缺乏对更高电压窗口的探索。最近,研究人员已经证明,体 SE 中的高电子传导性会加速氧化分解的动力学过程,这是导致严重枝晶晶粒生长的主要原因。许多研究都致力于通过添加电子导电率较低的第二相(如 SiO2、Al2O3、TiO2和 ZrO2)来解决干扰电子渗流问题。根据渗流理论,一旦填料的体积分数超过某个点,电子渗流就会被关闭,因此可以理解复合 SE 中电子电导率的下降。然而,这种策略的关键缺陷在于,体 SE 中的锂离子电导率会同步降低(尤其是在 RT 条件下),而且由于惰性填料的高硬度和亮度特性,机械性能也会下降。
还有一些学者尝试在 SEs 的晶界或电极界面中引入共价键分子和采用元素置换的方法,通过改变阳离子和阴离子形成离子/共价键无机化合物,这在一定程度上降低了 SEs 的电子传导性。尽管如此,复合 SE 的热稳定性、化学稳定性和电化学稳定性仍会显著下降,从而造成与锂阳极的界面不相容,导致自发反应和结构退化。
在无机 SE 中,LiBH4与金属锂相比具有独特的热力学稳定性,可保持低分子量、易变形和可压缩性,从而实现高能量密度和界面兼容性。然而,它们的氧化稳定性较差(相对于 Li^+^ /Li 氧化电压小于 2.0 V),并且在 125 ℃ 时 CCD 低至 2.8 mA cm^-2^,因此枝晶晶威胁严重。
知识要点 编辑本段
工作采用LiBH4和聚甲基丙烯酸甲酯(PMMA)之间的原位熔化反应(ISMR),在 SEs 颗粒表面的原位熔化反应层中生成共价键配位。这种配位在热力学上扩展了阴离子的氧化稳定性,在动力学上阻断了电子的渗流,起到了粘结剂的作用,将 SEs 粘结在一起,实现了优异的机械性能。因此,在较宽的工作温度范围内,可以实现前所未有的高电压工作稳定性和枝晶晶粒抑制。
ISMR 改性电解质的结构表征 ISMR 改性 SE 是通过与 γ-Al2O3、LiI 和 x wt~.% PMMA(x = 0,5)高能球磨 96 小时合成的,标记为 xPMMA。γ-Al2O3和LiI的引入是为了提高LiBH4~ SEs在RT下的锂离子传导性。
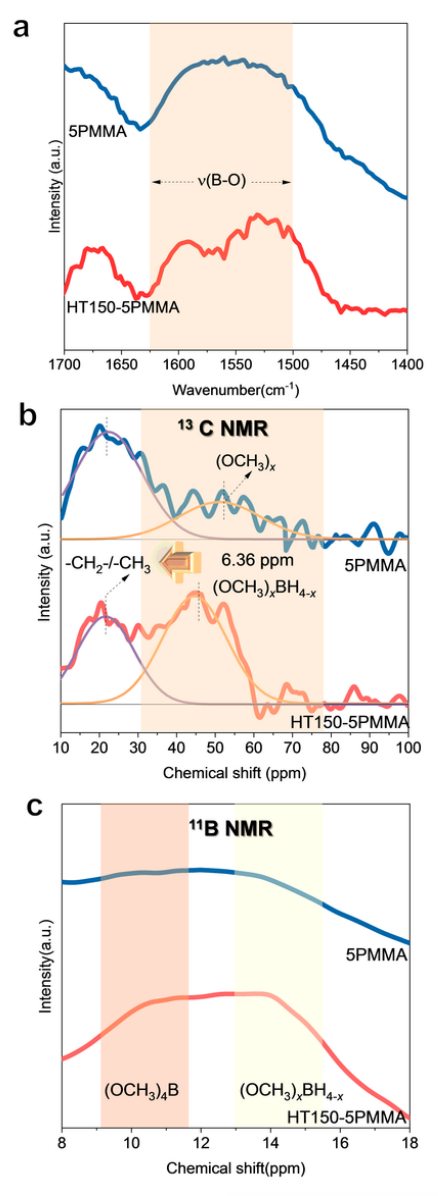
图 1 ISMR 改性 SE 的结构特征。
ISMR 改性电解质的稳定电压窗口和枝晶抑制能力
氧化稳定性差是 LiBH4面临的一大挑战。为了评估 HT150-0PMMA 和 HT150-5PMMA 的电压窗口,作者采用了三次连续循环伏安 (CV) 测试,从开路电压 (OCV) 到 10.0 V(相对于 Li^+^/Li),然后到 -0.2 V,再回到 OCV,扫描速度为 0.1 mV s^-1^。在第一个循环中,HT150-0PMMA 从 2.4 V 开始发生严重氧化,在 10 V 时达到 345.20 µA 的峰值电流(图 2a)。在 HT150-0PMMA 的后续循环中,氧化过程一直持续。在 HT1500PMMA 的第 1 个循环(图 2b 和 c)、第 2 个循环和第 3 个循环中,分别在 2.8、4.2 和 10.0 V 的截止电位下收集HT1500PMMA 的原位 B 1s 和 O 1s X 射线光电子能谱 (XPS) 谱。B1s XPS 谱(图 2b)中的原生 BH(185.94 eV)逐渐减小,伴随着高电压下氧化 BH4^-^(186.63 eV)的增加,表明氧化严重。第 2 和第 3 个周期的氧化分解情况较差。HT150-0PMMA 的 O 1s XPS 谱在三个循环中没有明显变化(图 2c)。HT1505PMMA 在前 3 个周期中的电压窗口出乎意料地高于 HT150-0PMMA,从 - 0.2 到 10.0 V(图 2d),氧化电流峰值为 0.93 µA,比 HT150-0PMMA 低 370 多倍。图 2e 和 f)、第 2 个和第 3 个周期 HT150-5PMMA 在 2.8、4.2 和 10 V 电压下的原位 B1s 和 O1s XPS 谱。图 2e 显示,在第 1 个循环中,氧化的 BH4^-^ 在 2.8、4.2 和 10.0 V 几乎可以忽略不计,在第 2 和第 3 个循环中也可以观察到类似的情况,这可以通过 XPS 定量得到证实。在第 3 个 CV 循环后,10.0 V 电压下的氧化 BH4^-^为 8.91 at%,比相同测试条件下的 HT150-0PMMA 低 6 倍。更有趣的是,在第 1 个周期(图 2f)、第 2 个周期(补充图 3f)和第 3 个周期(补充图 3h)的截止电压为 2.8、4.2 和 10.0 V 时,O 1s XPS 图谱中的 (OCH3)xBH4-x几乎保持不变,这表明 (OCH3)xBH4-x在高压操作下相当稳定。
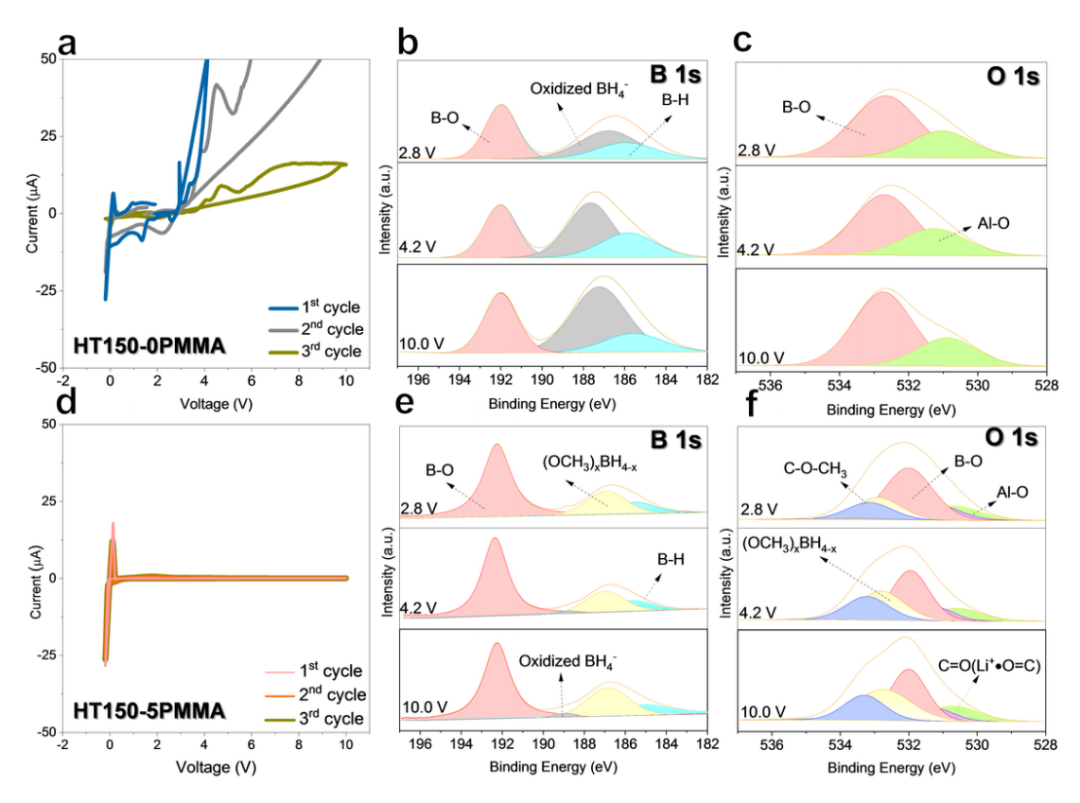
图 2 ISMR 改性 SE 的电化学稳定窗口。
8.0 ~ 15.0 V 的扩展 CV 操作表明,HT150-5PMMA 的上限电压窗口为 10.38 V,是所有基于 LiBH4的 SE 的最佳电压窗口的两倍。
为评估 HT150-0PMMA 和 HT150-5PMMA 的氧化稳定性,在较宽的温度窗口(-30 ~ 150 ℃)内进行了 17 次 CV 测试。HT150-0PMMA 在 -30、0、80 和 110 ℃ 出现不可逆氧化分解,在 130 ℃ 和 150 ℃ 直接失效。相比之下,HT150-5PMMA 在 -30、0、80、110、130 和 150 ℃ 下保持了 0 至 10.0 V 的高氧化稳定性,阴离子和氧化分解电流可忽略不计。
在 25 ℃ 下,HT150-5PMMA 中的 CCD(21.65 mA cm^-2^)是基于 LiBH4的 SE 中第一个也是最好的 CCD,是所有锂离子 SE 在 RT 下最佳 CCD(11 mA cm^-2^)的两倍。
HT150-0PMMA 和 HT150-5PMMA 的 CCD 在较宽的工作温度窗口中进行了测试(图 3f)。这一结果表明,HT150-5PMMA 在 -30 ℃ 至 150 ℃ 的温度范围内具有优异的枝晶抑制能力,在每个测试温度下的 CCD 都比 HT150-0PMMA 高 10 倍,特别的在超低温 -30 ℃(1.05 mA cm^-2^)和高温 150 ℃(342.00 mA cm^-2^)下。HT150-5PMMA 在宽工作温度窗口下的树突抑制表现出前所未有的适应性。
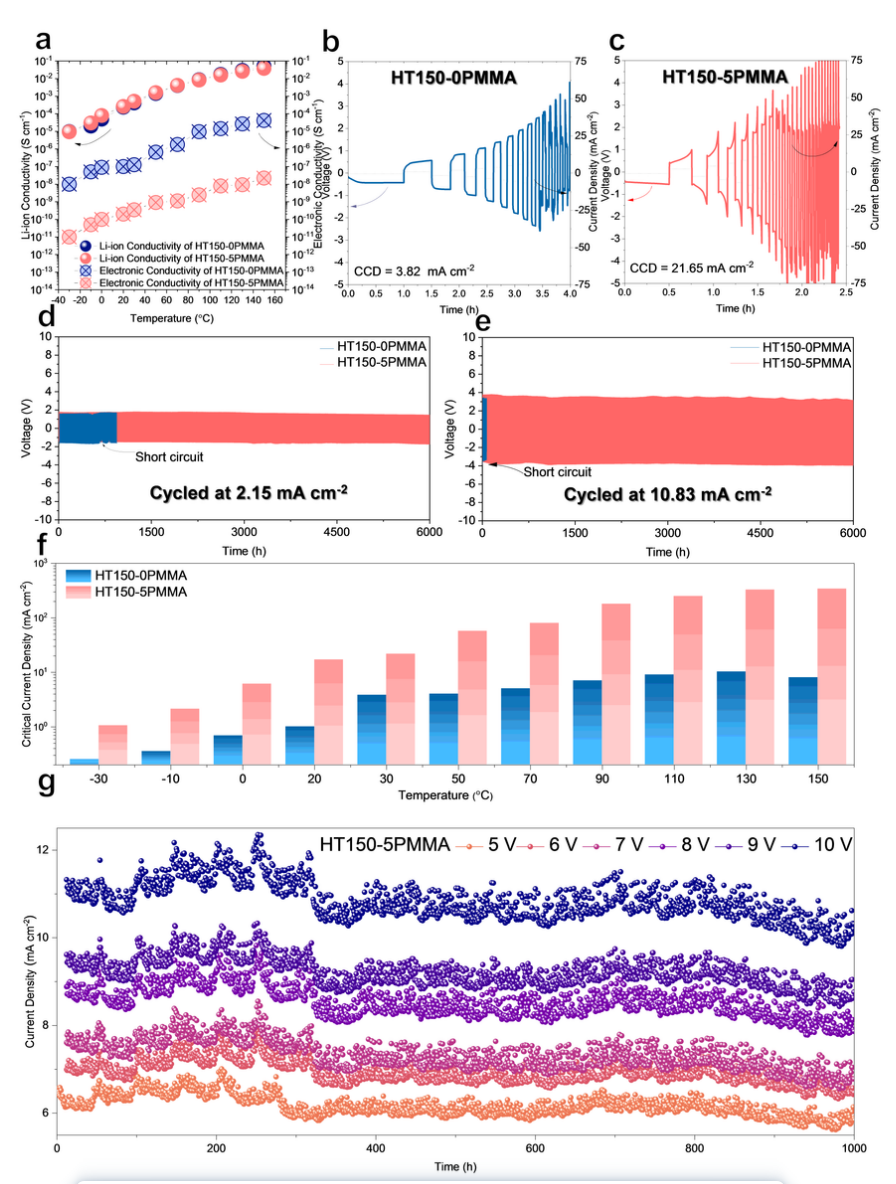
图 3 ISMR 改性 SE 的枝晶抑制。
通过第一原理密度泛函理论(DFT)计算,以揭示 PMMA 与 LiBH4之间的相互作用。计算了 LiBH4和 PMMA 构型的优化结构和分子静电势面映射。在图 5a 中,电子密度聚集在 BH4 阴离子(红色)周围。因此,在如此高的浓度下,局部电子趋于离域。根据计算,这种构型的第一电离能(从结构中失去一个电子所需的能量)为 4.81 eV。相反,随着 H^0^ 损失的增加,BHx^-^中的 B 可以牢固地与 -OCH3中的 O 配位(图 5b-e),从而导致整个构型中主要分布在 BH4^-^上的局部电子密度重新分布。因此,失去 1、2、3 和 4 个 H^0^后,LiBH4和 PMMA 的第一次电离能分别为 5.73、6.52、7.09 和 9.82 eV,这表明电子泄漏越来越难以从 BHx^-^ 中产生,从而表现出很强的电子局域性。
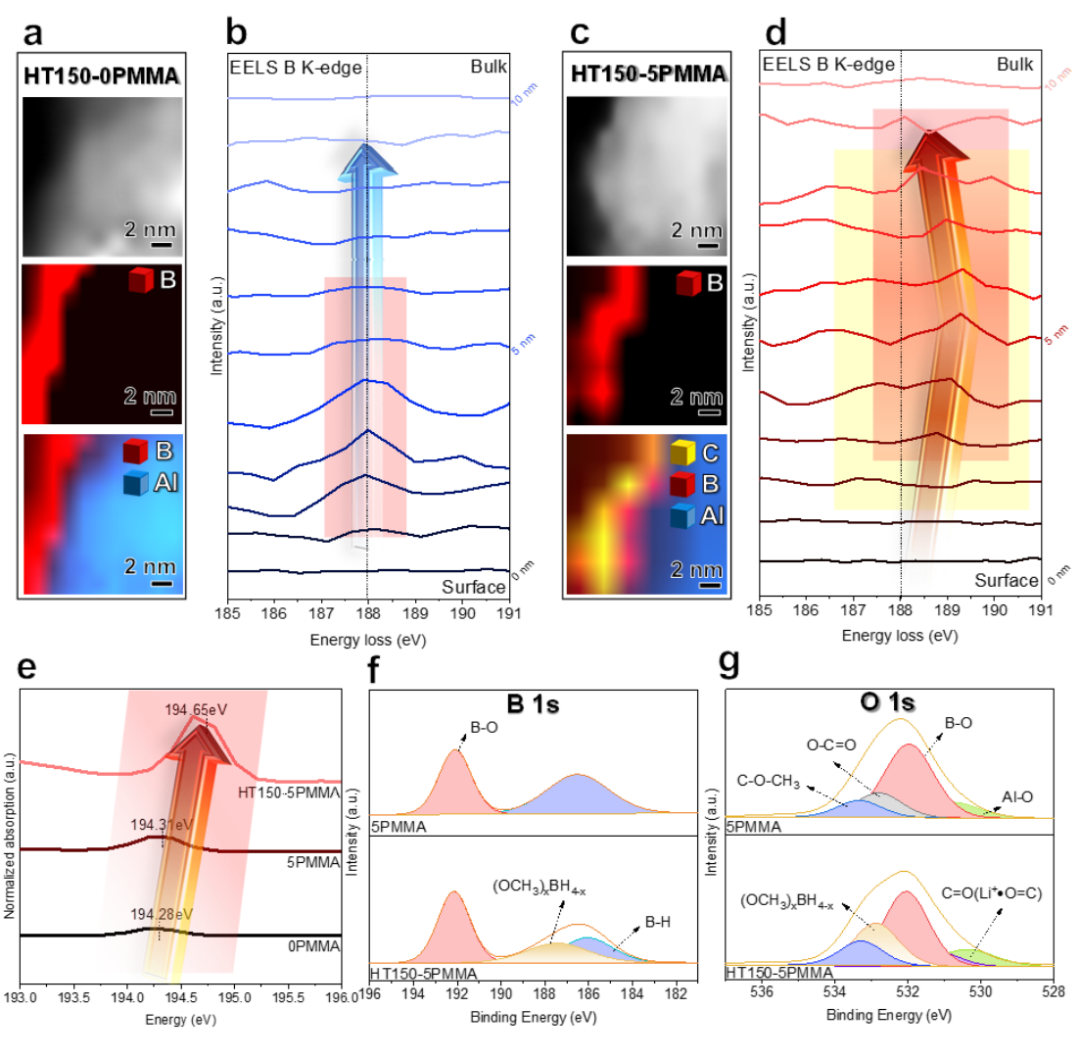
图 4 ISMR 改性 SE 中 (OCH3)xBH4-x的形成机制。
为了研究 (OCH3)xBH4-x在提高电化学性能特性方面的作用,根据 100 至 150 ℃ 的不同反应温度合成了一系列 HTy-5PMMA,以获得不同原子比的 (OCH3)xBH4-x。随后,测量了 (OCH3)xBH4-x、初始氧化电压、电子电导率和 CCD 之间的关系,如图 5f 所示。显然,随着 (OCH3)xBH4-x的增加,SEs 的初始氧化电压呈现出从 1.80 V 到 10.38 V 的线性增长,这证实了由于 (OCH3)xBH4-x在 SEs 中的强电子局域化,BH4^-^氧化过程中的电子交换可以在热力学上被关闭,这与 DFT 计算结果一致。此外,在图 5f 中,随着 (OCH3)xBH4-x的增加,电子传导性降低了几个数量级,这证实了由于 (OCH3)xBH4-x在块状 SEs 粒子表面的生成量增加,电子穿透可以被有效阻断。因此,CCD 呈线性增长,与电子导电率下降的趋势相反。因此,作者可以得出结论:氧化稳定性的增强和枝晶的抑制可归因于 ISMR 原位生成的 (OCH3)xBH4-x。
HT1505PMMA 在循环前以及循环 500 小时和 5000 小时后均未出现明显的裂纹、缺陷或锂沉积,这证实了其在长期镀锂和剥离过程中具有优异的结构完整性。
(OCH3)xBH4-x可作为 LiBH4SEs 颗粒表面的粘结剂,将 SEs 粘结成更紧凑的结构,从而大大增强了块体 SEs 的弹性行为,以适应循环过程中持续的应变应力释放。
通用研究 编辑本段
为了研究作者的发现的通用性,作者进一步测量了 Li2B12H12和 HT150-Li2B12H12-5PMMA 的电化学性能特征,它们是通过与 HT150-5PMMA 相同的反应合成的。结果表明,HT150-Li2B12H12-5PMMA在25 ℃时的CCD高达4.78 mA cm^-2^,是原生Li2B12H12的约6倍。在 LiAlH4和 LiNH2体系中也可以发现类似的趋势;不过,CCD 只增加了三倍。此外,所有经 ISMR 修饰的氢化物 SE 都能提供接近 10 V 的高度稳定的电压窗口,氧化电流相对于其原始形态几乎可以忽略不计。
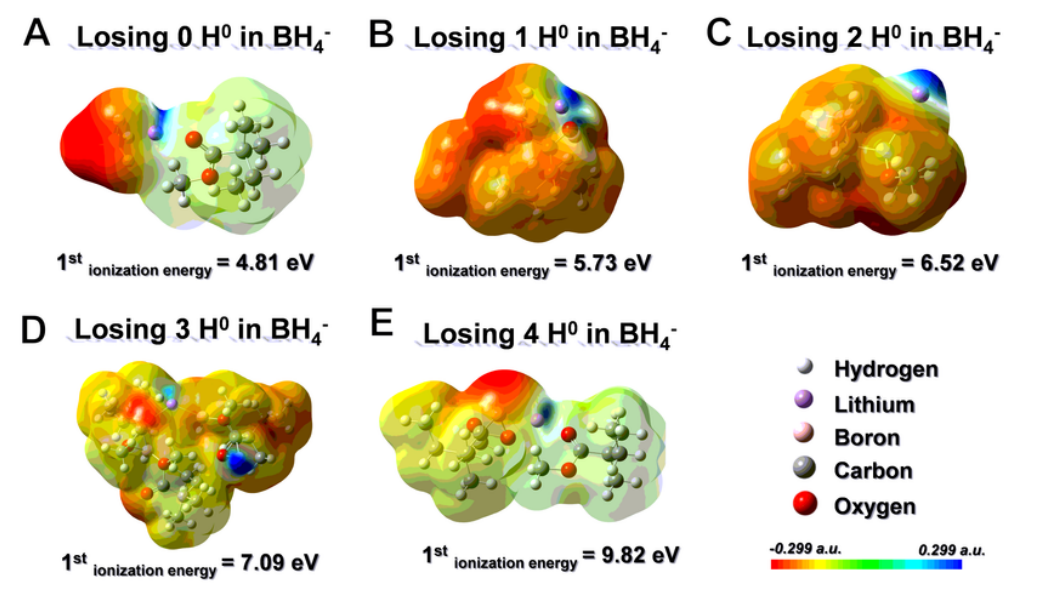
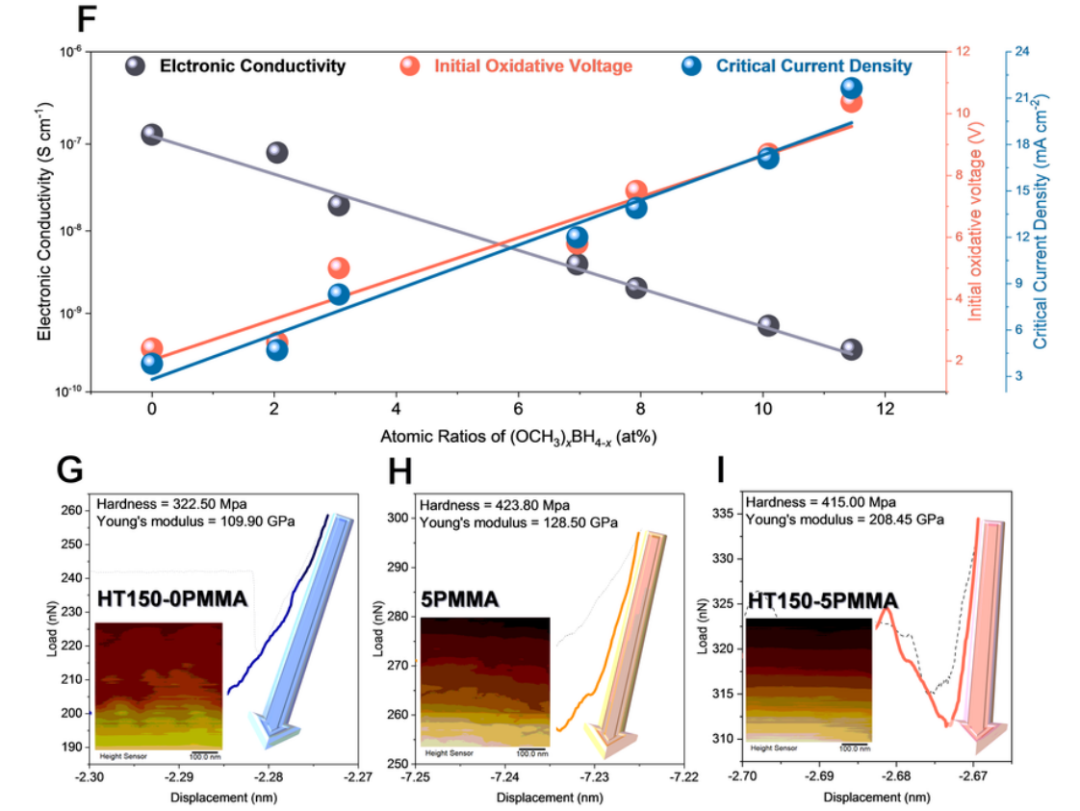
图 5 (OCH3)xBH4-x在提高电化学性能方面的作用。
使用 ISMR 改性电解质的高压锂金属电池
使用锂和钴酸锂作为电极,在 25 ℃ 下组装并测试了高压 ASSB。为了避免 LiBH4和 LiCoO2之间不可避免的反应,利用湿化学方法在 LiCoO2表面涂覆了 2 wt~.% 的 Li3InCl6~。使用 HT150-5PMMA 合成的 ASSB 初始比容量为 114.6 mAh g^-1^,在 0.1 C 下循环 100 次后的最高保持率为 95.8%(图 6a 和 b)。相比之下,使用 HT150-0PMMA 合成的 ASSB 的初始放电比容量较低,为 72.3 mAh g^-1^,随后容量迅速衰减,最终在 45 个循环后失效。
令人惊讶的是,含有 HT150-5PMMA 的钴酸锂 ASSB 实现了 111.4 mAh g^-1^ 的高初始放电容量,在工作电压为 3.0 至 4.2 V 的条件下,循环 200 次后的容量保持率为 100%,在 0.5 C 条件下循环 300 次后的容量保持率为 94.2%。
涉及 HT150-5PMMA 的高压钴酸锂 ASSB 在 3.0 至 4.2、4.6、4.8、5.0、8.0 和 10.0 V 的电压窗口中进一步循环。充放电曲线和相应的循环性能特征分别如图 6e 和 f 所示。作者的 HT150-5PMMA 在 3.0 至 4.6、4.8 和 5.0 V 的电压范围内具有较高的可逆性,在 25 ℃(0.1 C)条件下的可逆初始放电容量分别为 128.7、135.0 和 144.3 mAh g^-1^。然而,由于钴酸锂在高 SOC 下过度脱锂导致结构不可逆崩溃,45 个 ASSB 循环一次,在 8 V 和 10 V 的上限截止电压下分别显示 130.5 和 136.0 mAh g^-1^ 的初始放电容量,这表明 ISMR 改性 SE 在高压锂金属 ASSB 中大有可为。
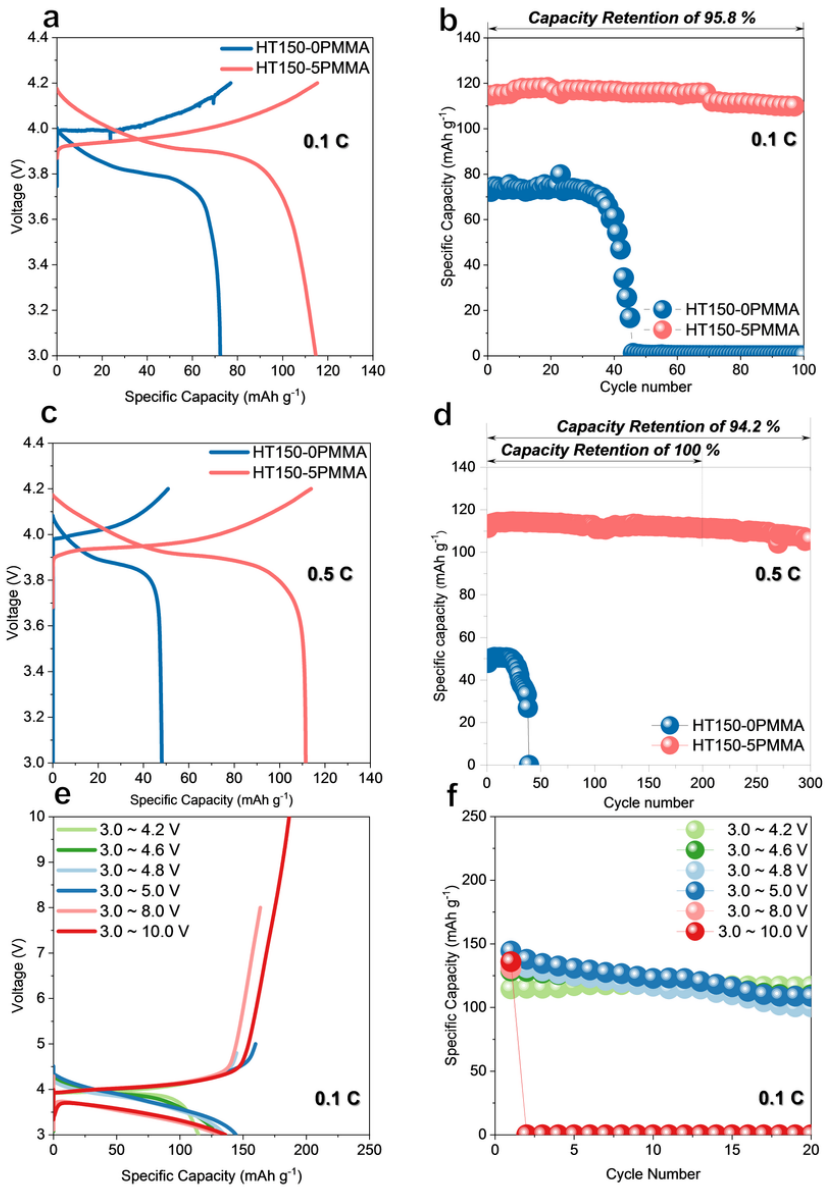
图6 使用 ISMR 改性 SE 构建的高电压Li-LCO ASSB。
反应策略 编辑本段
工作开发一种新颖的原位熔化反应策略,在 LiBH4SEs 的颗粒表面生成共价键配位,以解决 SEs 氧化稳定性差和枝晶问题。这种配位从热力学角度提高了阴离子的内在氧化稳定性,并从动力学角度阻止了 SEs 粒子表面的电子传导,从而抑制了枝晶的生长。此外,它还起到粘结剂的作用,有助于在锂的连续沉积和剥离过程中实现出色的机械性能,以适应应力应变的释放。因此,所获得的 SE 保持了创纪录的高电压窗口(0 ~ 10 V),峰值氧化电流比同类产物低 370 倍;此外,在 25 ℃ 下具有前所未有的 CCD(21.65 mA cm^-2^),在 10.83 mA cm^-2^ 下具有 6000 小时的超长循环稳定性,在 10 V、25 ℃ 下具有 1000 小时的超长循环稳定性,实现了从 - 30 ℃ 到 150 ℃ 的宽工作温度窗口。他们的钴酸锂电池在 3.0 至 4.2 V、60 mA g^-1^ 的条件下循环 200 次后,容量保持率达到 100%,并且在 3.0 至 5.0 V 范围内具有可逆循环稳定性。作者的发现为促进高能量密度 ASSB SE 的电化学稳定性提供了一个清晰的视角。
附件列表
词条内容仅供参考,如果您需要解决具体问题
(尤其在法律、医学等领域),建议您咨询相关领域专业人士。